

ORIGINAL PAPER
Xanthine oxidase targeted model setup and its application for antihyperuricemic compounds prediction by in silico methods
1
School of Food Sciences and Engineering, South China University of Technology, China
2
China-Singapore International Joint Research Institute, China
Submission date: 2022-01-26
Final revision date: 2022-02-25
Acceptance date: 2022-03-01
Online publication date: 2022-03-23
Publication date: 2022-03-23
Corresponding author
eFood 2021;2(6):296-306
KEYWORDS
hyperuricemiamolecular dockingMultiple linear regressions (MLR)Principal component regression (PCR)and Artificial neural network (ANN)
TOPICS
ABSTRACT
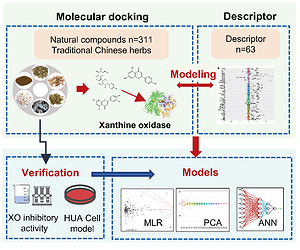
To achieve potential alternatives for hyperuricemia therapeutics, a novel structure-docking energy relationship model was established for high-throughput screening inhibitors of xanthine oxidase (XO). Molecular docking was performed between XO and 311 natural compounds from 6 traditional Chinese herbs. Then, structure-docking energy relationship model was simulated between molecular docking energy and 63 molecular descriptors by multiple linear regressions (MLR), principal component regression (PCR), and artificial neural network (ANN), respectively. The results showed that the ANN model was the best model to predict the docking energy of XO with the coefficient of determination (R2) and mean squared error (MSE) at 0.8746 and 0.9414, respectively. The data of XO inhibitory activity were consistent with the prediction in vitro, which was also further confirmed by hyperuricemia cell model. The results suggested that the structure-docking energy relationship model provides a paradigm framework for the screening of XO inhibitors.
| eISSN: | 2666-3066 |
We process personal data collected when visiting the website. The function of obtaining information about users and their behavior is carried out by voluntarily entered information in forms and saving cookies in end devices. Data, including cookies, are used to provide services, improve the user experience and to analyze the traffic in accordance with the Privacy policy. Data are also collected and processed by Google Analytics tool (more).
You can change cookies settings in your browser. Restricted use of cookies in the browser configuration may affect some functionalities of the website.
You can change cookies settings in your browser. Restricted use of cookies in the browser configuration may affect some functionalities of the website.